|
|
If I can't calculate it, I don't understand it
Richard Feynman
 With the advent of ever increasing compute power also comes the challenge of developing realistic models and empirical potentials to simulate physical and
chemical processes that accurately describe experiment. Molecular-dynamics (MD), Monte Carlo (MC), Kinetic Monte Carlo (KMC) and phase-field
simulations provide access to the kinetic behavior of these alloys, bridging system studies with DFT to understand the effects of poly-domain multicomponent
alloys with well-defined microstructure, compositionally gradients, strain fields, and interfaces. Effects of induced charges on metal surfaces and catalytic reactivity
have been quantitatively explored in combination with first principles methods and experimental measurements, and new insights into oxide surface thermodynamics
and hydration reactions have become feasible in high accuracy. Indeed KMC simulations have clearly shown that classical models for alloy stability in for example
Ni-Al-Cr alloys are insufficient; they have been able to explain the microscopic mechanisms leading to phase separation and decomposition of the multicomponent system,
yet still employed empirical fits to diffusion barriers. More recently it has become clear that unit cell level interactions, including electronic structures,
spin states, and phonon modes must be accurately captured to provide realistic energy barriers.
With the advent of ever increasing compute power also comes the challenge of developing realistic models and empirical potentials to simulate physical and
chemical processes that accurately describe experiment. Molecular-dynamics (MD), Monte Carlo (MC), Kinetic Monte Carlo (KMC) and phase-field
simulations provide access to the kinetic behavior of these alloys, bridging system studies with DFT to understand the effects of poly-domain multicomponent
alloys with well-defined microstructure, compositionally gradients, strain fields, and interfaces. Effects of induced charges on metal surfaces and catalytic reactivity
have been quantitatively explored in combination with first principles methods and experimental measurements, and new insights into oxide surface thermodynamics
and hydration reactions have become feasible in high accuracy. Indeed KMC simulations have clearly shown that classical models for alloy stability in for example
Ni-Al-Cr alloys are insufficient; they have been able to explain the microscopic mechanisms leading to phase separation and decomposition of the multicomponent system,
yet still employed empirical fits to diffusion barriers. More recently it has become clear that unit cell level interactions, including electronic structures,
spin states, and phonon modes must be accurately captured to provide realistic energy barriers.
 Our team is uniquely positioned to take the leading role in addressing our main research objectives as the assembled materials theory/simulation team has already made substantial
contributions to understanding kinetics and redox chemistry, transition metal oxides, and microstructure evolution at multiple length and time scales.
A wide range computational approaches will be applied to address the challenges. Active interactions among the theory/modeling PIs, in
additional to the experimentalists, is required to build the required local corrosion and oxidation understanding. Broadly, the elucidation of nucleation kinetics, barrier heights, and
transport parameters will be achieved using the force fields (Heinz) in combination with advanced DFT methods which will provide interface- and local composition dependent energetics
(Rondinelli & Marks). Such parameters can then be used for phase field models to capture the dynamics on the micrometer scale by the Voorhees group.
Our team is uniquely positioned to take the leading role in addressing our main research objectives as the assembled materials theory/simulation team has already made substantial
contributions to understanding kinetics and redox chemistry, transition metal oxides, and microstructure evolution at multiple length and time scales.
A wide range computational approaches will be applied to address the challenges. Active interactions among the theory/modeling PIs, in
additional to the experimentalists, is required to build the required local corrosion and oxidation understanding. Broadly, the elucidation of nucleation kinetics, barrier heights, and
transport parameters will be achieved using the force fields (Heinz) in combination with advanced DFT methods which will provide interface- and local composition dependent energetics
(Rondinelli & Marks). Such parameters can then be used for phase field models to capture the dynamics on the micrometer scale by the Voorhees group.
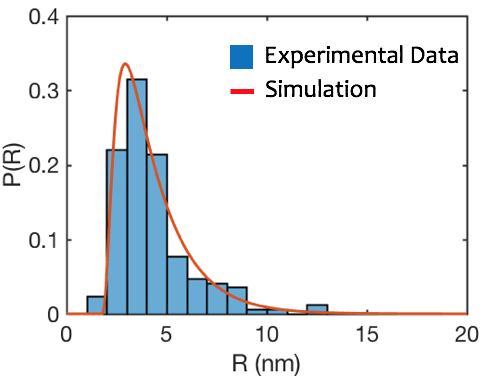 In addition to phase-field modeling, the Voorhees group is developing mean-field models to describe the nucleation and growth of oxide islands during the initial stages of oxidation. Working in close collaboration with the experimental groups, we are also beginning to apply various levels of theory and computation to the problems of solute trapping and instabilities during oxidation/corrosion processes.
In addition to phase-field modeling, the Voorhees group is developing mean-field models to describe the nucleation and growth of oxide islands during the initial stages of oxidation. Working in close collaboration with the experimental groups, we are also beginning to apply various levels of theory and computation to the problems of solute trapping and instabilities during oxidation/corrosion processes.
The figure to the right shows a comparison of the results of a simulation of island growth on a NiCr0.05 alloy to experiment. The simulation describes how the island size distribution changes as islands grow using data from early oxidation steps as the initial condition.
|
